
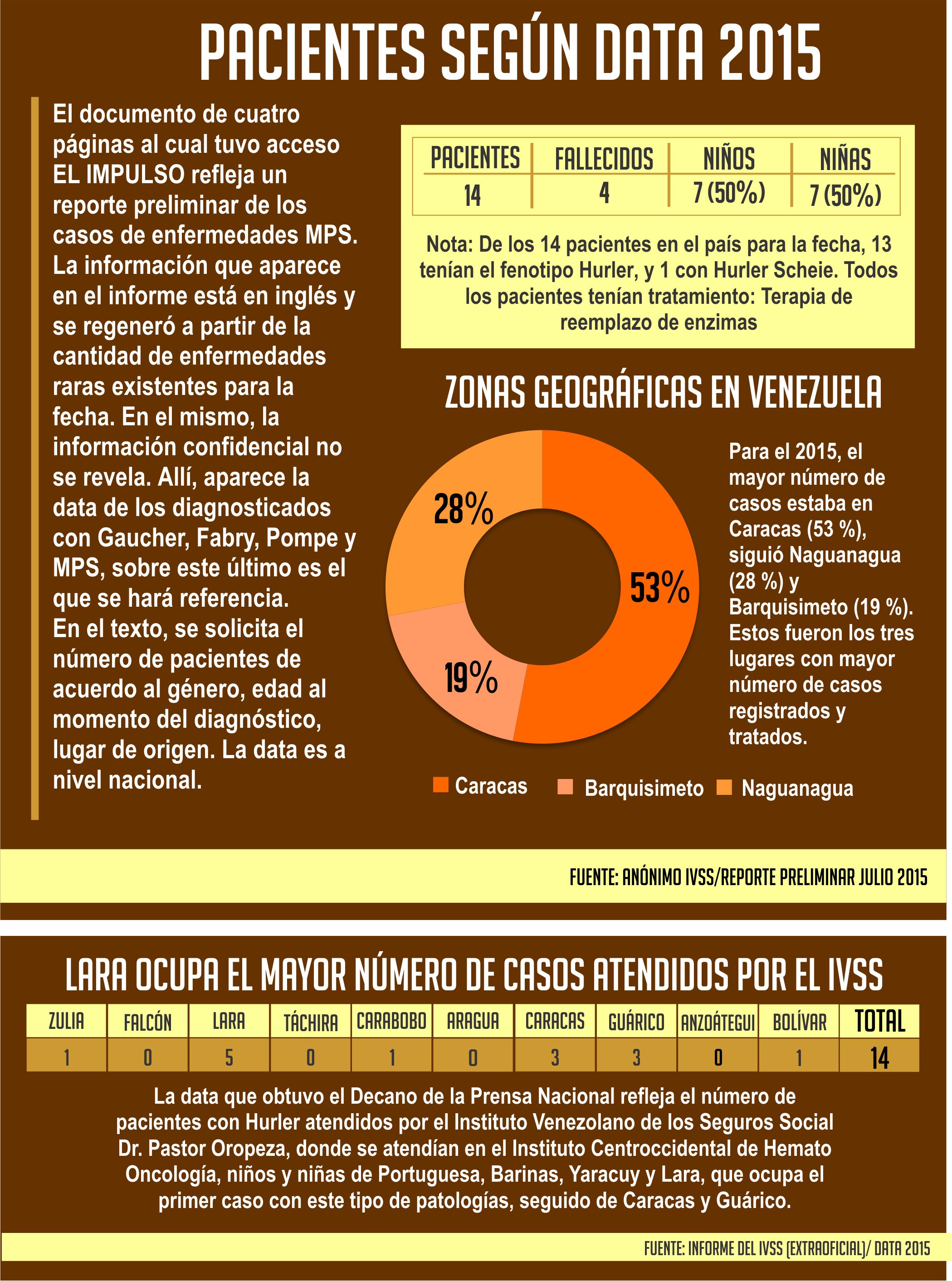
La emergencia humanitaria compleja en Venezuela es cada vez más evidente en los sectores vulnerables de la población, en especial aquellos que integran el grupo de enfermedades raras. Los pacientes registrados en el Instituto Venezolano de los Seguros Sociales (IVSS), con padecimiento de depósito lisosomal mucopolisacaridosis tipo I Hurler (MPS I), aún esperan el medicamento (enzima artificial) de alto costo Aldurazyme (Laronidasa), que solo el Estado puede adquirir a través del laboratorio Genzyme Sanofi, una trasnacional biotecnológica estadounidense con sedes en más de cien países en las principales capitales del mundo.
De acuerdo a informes obtenidos por EL IMPULSO, en el IVSS Dr. Pastor Oropeza de Barquisimeto, estado Lara, que recibe a pacientes del Centroccidente y Los Llanos venezolano (Lara, Yaracuy, Portuguesa y Barinas), la cifra hasta agosto de 2018 registra dos niños fallecidos por falta de medicamentos y otros cinco esperan el Aldurazyme desde enero de 2017. Solo una niña de los ocho casos, pereció por supuesta mala praxis médica hace un año con seis meses (ella recibía tratamiento en ese entonces).
La compañía farmacéutica situada en Miranda (Urbanización Boleíta Norte, prolongación calle Vargas con segunda Trasversal, municipio Sucre), por los momentos no ha recibido el pedido por parte del Ministerio del Poder Popular para la Salud (MPPS), con el propósito de satisfacer la demanda de pacientes registrados, no solo de esa patología, sino de Gaucher, Fabry o Pompe, otras de las enfermedades afectadas por la escasez de medicamentos en Venezuela, que suman 137 casos en todo el territorio nacional, sin contabilizar los fallecidos. En esos pacientes, no se incluyen aquellos, cuyos padres todavía evitan ir al centro asistencial a dar a conocer el caso que les preocupa, porque saben cómo la situación país los afecta directamente. De allí, que sus esperanzas están quebrantadas.
El Aldurazyme sirve como sustituto enzimático de los pacientes con Hurler I, que por problemas genéticos no producen la L-iduronidasa (enzima), encargada de procesar los glucosaminoglucanos (azúcar), lo que genera acumulación de tejidos en el cuerpo y la presencia de síntomas como enanismo, retraso del desarrollo motor (movilidad de las extremidades) y mental, así como deformidades del esqueleto, cara plana y frente arqueada. No se encuentra en farmacias, sino a través de instituciones encargadas de la investigación genética en el mundo. A la fecha, autoridades del IVSS y del MPPS no se han pronunciado al respeto.
En Venezuela la Asociación Civil Humberto Da Silva (Achds) y la Asociación Venezolana de Pacientes con Enfermedades Lisosomales (Avepel), se encargan de hacerles seguimiento a estos pacientes que requieren de sus medicamentos para poder mantenerse.


Avepel: “Es urgente que lleguen los tratamientos”

Gregoria Sameco, representante de Avepel y paciente de Gaucher, comentó sobre la situación que les aqueja a los pacientes con enfermedades raras en varios estados de Venezuela.
Manifestó que todo comenzó con una demanda en contra del IVSS, que hicieron algunos para obtener el medicamento. “Se ganó la lucha y solo recibieron el tratamiento los pacientes asegurados. A partir del 2009 el resto de quienes padecíamos las patologías lisosomales comenzamos a recibir el beneficio de manera gratuita y obligatorio”, dijo.
Sameco explicó que el tratamiento, según el registro que ellos llevan, no llega al Seguro Social a mediados de 2016. Un año después, se recibieron las últimas infusiones a través de ayuda internacional, pero para personas específicas que tenían un estado de salud más delicado.
Hizo un llamado a las autoridades competentes, quienes son los responsables de hacer la solicitud para que llegue el tratamiento de alto costo a Venezuela. “Tenemos derecho a la salud, es un derecho legal, si no recibimos nuestro tratamiento nuestra calidad de vida se va a deteriorar cada vez más, e incluso llegar a la muerte. Como venezolanos y pacientes de estas patologías hacemos un llamado al IVSS y al Gobierno nacional, para que suministren nuestro tratamiento”, sentenció.[/vc_message]



Marlyn Roldán Valencia sigue luchando por su hija. A tan solo 20 minutos de Barquisimeto está la capital del municipio Jiménez, Quíbor (Urbanización La Ceiba), donde vive una valiente mujer de 32 años que hoy lucha por darle las mejores condiciones de vida a su hija de 10 años de edad con síndrome de Hurler I, que tampoco recibe tratamiento con Aldurazyme.
Aunque vive muy de cerca la crisis humanitaria compleja, mantiene la esperanza de que su adorada hija obtenga nuevamente la enzima artificial que le falta. No se da por vencida: eleva su voz adonde crea conveniente y aprovecha cualquier espacio para mostrar su caso, que hoy es reflejo de políticas públicas sanitarias poco eficientes.
Ha hablado con médicos, fundaciones y hasta tuvo cercanía con políticos –luego de falsas promesas-, pero no recibe una respuesta satisfactoria. Ni siquiera el laboratorio Genzyme Sanofi podría ayudarle a través de la fundación ICAP (International Charitable Access Program), porque solo cubre a niños hasta los cinco años de edad.
Se mantiene preocupada y siempre alerta ante el estado de salud de la niña que, a pesar de estar consciente de su enfermedad, hace el esfuerzo diario por mantenerse de pie, sonriente y con un gesto de amor hacia quienes están cerca de ella.
El tratamiento llegaba con intermitencia al Pastor Oropeza
La niña fue diagnosticada hace ocho años con Hurler I, en el Hospital Universitario Antonio María Pineda (HUAMP), por el doctor Pedro Estrada, genetista y profesor de la Universidad Centroccidental Lisandro Alvarado (UCLA), quien la refirió a la Unidad de Hemato Oncología del Hospital Pastor Oropeza de Barquisimeto, donde sería incluida en un proyecto para pacientes con enfermedades raras.
Cuando tenía dos años de edad, la pequeña inició el tratamiento por un lapso de seis meses, todas las semanas, comentó Roldán Valencia. En ese momento, “sufrió cambios físicos: perdió los vellos de la piel y de la cabeza, pero progresivamente comenzaron a salir sin tanta abundancia. Sus rasgos cambiaron mucho”.
Mientras recibía el remplazo de enzima, presentaba mejor movilidad en la articulares, aunque con poca dificultad para caminar. “Hizo su vida normal durante seis años completos que duró continuamente en el Pastor Oropeza recibiendo tratamiento. Pero en el 2016 comenzó una falla de tratamiento progresivo por meses, hasta que dejó de llegar”, expresó la mamá.
Pacientes de otras patologías son prioridad para el Estado
Entre excusas y sin garantía del derecho a la vida garantizado en la Constitución de Venezuela, Roldán Valencia sigue con energía para exigir el medicamento que como venezolana le corresponde a su pequeña y es responsabilidad del Estado. Nadie le ha dado alguna explicación que justifique los motivos por los cuales la enzima no llega al país, solo ha escuchado que por ser una medicamento de alto costo, existe dificultad para la importación. “Nos dijeron que la adquisición era imposible, porque habían muchos pacientes oncológicos que necesitaban tratamiento y era el mayor número de personas.
La demanda de ellos era más fuerte que la de nosotros. En ese entonces eran cuatro niños, pero después se unieron otros hasta alcanzar el número de 8 pacientes (del Centroccidente) y evitar que se perdiera el tratamiento”, comentó.
En la actualidad, las condiciones de su niña no son las adecuadas para ir a la escuela, donde compartía con sus compañeros de clases y recibía una educación apta en medio de sus circunstancias.
La enzima desapareció por completo
Familiares de pacientes con síndrome de Hurler I saben que el Estado rompió convenio con el laboratorio Genzyme Sanofi situado en Miranda y que las respuestas positivas escasean. Incluso, se les informó que en caso de que llegara el tratamiento, debían registrar nuevamente los datos del paciente para iniciar el proceso desde cero:
“Eso implica firmar los permisos y las autorizaciones para obtener el medicamento”, explicó Roldán Valencia, quien en noviembre de 2017 fue hasta el IVSS en Caracas, donde le dijeron que “la compra era en divisas y no contaban con las necesarias para tal fin”.
“Me reuní con la secretaria del doctor y dijo que el problema era institucional, gubernamental”, expresó. Por lo que le recomendaron que hiciera presión a través de la política partidista, una estrategia que poco querían implementar.
“Nos dijeron que esperáramos que la actual gobernadora de Lara, Carmen Meléndez, afianzara su proyecto, que ella nos iba a ayudar. Incluso, nos pusimos en contacto con ellos por medio de Avepel, y nos pidieron que participáramos en eventos mientras estaba en campaña electoral”, pero todo fue en vano, nadie le respondió, la engañaron a ella y a otras madres preocupadas por la situación de sus hijos.
Roldán Valencia manifestó que asistieron a tres encuentros políticos, pero en ninguno de ellos se logró hablar con la máxima autoridad regional.
“Solo se recibió un escrito, en el cual se explicaba la enfermedad de los niños y de la necesidad de que volviera el medicamento. Si ellos no lo reciben, progresivamente va a ir empeorando su cuadro clínico hasta llegar al punto de morir por falta de medicamentos”.
Entre el dolor, la angustia y la esperanza
La joven Roldán Valencia sabe cómo está la situación país y la distribución de medicamentos de alto costo en los hospitales pertenecientes al Instituto Venezolano de los Seguros Sociales (IVSS). Solo se aferra al impacto de las redes sociales para encontrar respiro a la falta de tratamiento de su hija y la de otros pacientes que están en riesgo de morir.
En medio de lágrimas y la escucha activa de su hija mientras se realizaba la entrevista, pidió mayor consciencia sobre la importancia del Hurler I en Venezuela. “Mi hija está mal, su salud ha desmejorado mucho. Ella era una niña muy activa, que asistía a la escuela. Tenía buena relación con sus compañeros. Hoy en día, camina con dificultad, amanece cansada, no quiere caminar ni jugar. A nivel respiratorio está muy comprometida”, indicó.
Recientemente la niña de 10 años estuvo hospitalizada en el área de Pediatría del Pastor Oropeza, con un asma crónica no controlada. Incluso, comentó que el bazo e hígado siguen en aumento, eso le comprime el diafragma y las dificultades respiratorias se acentúan.
“Ha sido fuerte…”, expresó con profundo dolor. “Ella todos los días me pregunta cuándo va a iniciar el tratamiento, porque sabe que está consciente de que el medicamento le ayuda a estar mejor, a sentirse bien. ¿Qué explicación le doy? Cómo le hago entender que no es algo que no quiero darle o que los doctores no quieren darle, sino que lamentablemente con la situación país es difícil de adquirirlos”.
Ella y otras madres preocupadas han hecho lo posible por ser escuchadas. En algún momento participaron en un programa de televisión transmitido por el canal regional Promar, con el propósito de visibilizar la situación. En el espacio televisivo estuvo otra niña de Barquisimeto, que murió dos semanas después de haberle realizado la publicación audiovisual.

En el caserío Los Cerritos de la parroquia Tintorero, municipio Jiménez, está el caso de una niña de 5 años de edad. Su mamá, Amanda Ruiz, se aferra a Dios en medio de la angustia que le genera saber que su hija en cualquier momento puede morir. Tampoco tiene tratamiento, siendo su garantía responsabilidad del Estado venezolano.
Las expectativas de vida de la pequeña disminuyen ante la falta de enzima artificial de alto costo. “Se ha enfermado mucho”, explicó Ruiz. Ha tenido constantes vómitos y debilidad en las articulaciones, así como dificultad respiratoria. “El doctor le ha sacado mucha flema, pero sigue igual”.
Sus órganos están inflamados, solo el Aldurazyme puede mejorar la calidad de vida, más no curarla. “Le pido mucho a Dios para que me ayude… hemos llorado mucho aquí en la casa. No es justo lo que estamos viviendo”, indicó. Teme que su hija muera de un paro respiratorio como ha ocurrido con casos anteriores.
Igual diagnóstico tiene un niño de 6 años de edad. Él vive con su mamá en Acarigua, la señora Nayalí Hidaldo. Cuando tenía dos años de edad comenzó a recibir el tratamiento, pero ante la falta del mismo, se ha complicado, presentando problemas respiratorios.
Su calidad de vida ha desmejorado. Antes jugaba bicicleta, “gracias a Dios todavía camina y está activo”, dijo.
Todos los niños diagnosticados con Hurler en el Centroccidente están desesperados ante la ausencia de la enzima artificial que les controla el avance de la enfermedad.

Yaquelín Gómez despidió a su hija en La Batalla, al oeste de Barquisimeto, y aún la recuerda con mucho dolor. No olvida los gestos de su niña de 10 años cuando aún vivía.. Si bien ya no está entre su núcleo familiar físicamente, siempre la tienen presente y por eso, seguirá siendo portavoz de aquellos que requieren el Aldurazyme para sobrevivir.
Yaquelín del Carmen Gómez Garrido contó que “su tesoro” nació sin movilidad, pero cuando inició el tratamiento vio mejoría. Lamentó que en la farmacia del Seguro Social Pastor Oropeza, le dijeran con frecuencia que el medicamento no iba a llegar. “Me tuvieron como un mes diciéndome que pasara entre semana para ver si nos daban respuestas… y así nos tenían, hasta que mi niña murió”, reveló.
Fue referida al IVSS por el doctor Pedro Estrada, de la Unidad de Genética Médica de la UCLA, el 2 de junio de 2009, diez meses después de haber nacido. La insuficiencia respiratoria era frecuente en la niña, hasta el punto de quitarle la vida en el Hospital Universitario de Pediatría Dr. Agustín Zubillada (HUPAZ), donde permaneció por cuatro días hospitalizada. Allí, dejó de existir producto de una neumonía.
El otro paciente que murió a la espera del medicamento vivía en sector El Calvario de Quíbor, en una vivienda construida de barro y en medio de una familia que le robaba sonrisas, junto a sus dos hermanos. María Gil y su esposo Jeison Daza, contaron que el niño de 8 años de edad, falleció por un paro respiratorio.
“Cuando tenía un año y medio comenzó a recibir la dosis, gota por gota”, dijo Daza, quien recordó que antes de partir su pequeño, habían transcurrido 6 meses del último tratamiento.
Su diagnóstico se realizó cuando tenía tres años de edad, gracias a la curiosidad de su abuela, quien mientras iba en un ruta, vio que otro niño presentaba características similares a las de su pariente. De allí, que comenzó la alerta y lo llevaron al pediatra para ser diagnosticado con Hurler I. “Nosotros no conocíamos esa enfermedad. Nadie nunca nos dijo nada”, explicó Gil.

El doctor Pedro Estrada, con especialidad en Anatomía Patológica, maestría en Genética y doctorado en Ciencias Médicas, además de profesor titular de la Universidad Centroccidental Lisandro Alvarado (UCLA), explica en qué consiste la enfermedad, esa que ha tocado tratar en el estado Lara, donde se encuentra el mayor número de casos del país.
En Barquisimeto, aseguró, existe la MPS I, MPS II, y otro caso con MPS IV (Síndrome de Morquio), así como una sospecha de MPS III que es el Sanfilippo.
 Sobre el Hurler I, aseguró que desde el punto de vista enzimático, “el glucosaminoglucanos se debe metabolizar para ser excretado de forma adecuada, a través de la L-iduronidasa, el cual no se produce. De manera que se va almacenando en ciertos órganos blancos y provoca la enfermedad”.
Sobre el Hurler I, aseguró que desde el punto de vista enzimático, “el glucosaminoglucanos se debe metabolizar para ser excretado de forma adecuada, a través de la L-iduronidasa, el cual no se produce. De manera que se va almacenando en ciertos órganos blancos y provoca la enfermedad”.
-¿Cuáles son los órganos más afectados cuando existe la acumulación del glucosaminoglucanos?
-Generalmente el hígado y el bazo van a aumentar de tamaño, así como ocasionar rigidez articular, por eso vemos en ellos una mano en garra, que le va a impedir los movimientos de flexo extensión. Incluso, se almacena a nivel de la columna, por eso presentan una cifosis (especie de giba) y problemas osteoarticulares del hueso propiamente. Lo más delicado de la condición es la presencia del glucosaminoglucanos en el sistema nervioso central, por lo que el paciente va perdiendo progresivamente unas funciones que había adquirido.
-¿En qué momento comienzan a presentarse los síntomas?
-El cuadro clínico inicia desde los seis a siete meses de haber nacido, porque mientras tanto se ha venido manejando con la enzima que la madre le previó a través del cordón umbilical y la misma no pudo metabolizar el glucosaminoglucanos por cierto tiempo.
-¿Cuáles son las formas clínicas de la mucopolisacaridosis tipo I?
-Existen tres formas: el Hurler clásico, que se presenta en los primes meses de haber nacido; Hurler-Sheie, que es una forma intermedia, en el cual todo va a ocurrir pero tardíamente; y la Sheie, que es la forma leve, en pacientes que pueden llegar a la vida adulta sin tratamiento, porque los glucosaminoglucanos también producen espesamiento de las secreciones (moco espeso), lo que va a provocar problemas respiratorios.
-¿Cuánto puede vivir un paciente sin tratamiento?
-En el historial clínico natural de la enfermedad, después de los 15 años fallecen, porque van perdiendo progresivamente sus funciones. Ellos tienden a presentar cuadros como si fueran de demencia, falta de movilidad, hasta que mueren. En las etapas finales presentan convulsiones por su propia acumulación de glucosaminoglucanos en el sistema nervioso central. En el caso del Sheie, no tiene esta afectación general.
-¿Por qué es importante el tratamiento y cómo fue ese proceso de llegada del medicamento al país?
-Es supremamente importante. Hasta hace cuatro años, y en eso aunque suene un poco egocéntrico, no por iniciativa mía, sino de los padres con paciente MPS I y MPS II, hicimos en 2006 una campaña fuerte para que llegara el medicamento en Venezuela. De allí que el IVSS a través de la Unidad de Medicamento de Alto Costo la canalizó. Se hizo el contacto con varias empresas para la MPS II y utilizando la igualdad, se logró para la MPS I, porque ya los pacientes con Gaucher recibían el tratamiento.
Se logró que el Gobierno contactara a la empresa internacional Shire, que lo estaba colocando en Argentina y Brasil, y sirvieron de intermediario. Vinieron para acá e hicieron que se comprara el medicamento en el año 2006.
En un principio se hizo de manera directa. Es decir, el Estado lo compraba a Shire, llegaba al IVSS y este se encargaba de enviarlo a los centros regionales que tuviesen pacientes, cumpliendo una cadena frío.
En el caso de la MPS I, el tratamiento se consiguió un poquito más tarde, en 2008.
-¿Cómo es la expectativa de vida con tratamiento?
-Mejora mucho, porque los síntomas y los signos viscerales, el aumento del hígado, del bazo y rigidez articular, mejora mucho.
-¿Qué desventaja tenía el medicamento?
-El gran problema del medicamento es que no atraviesa la barrera hematoencefálica que tenemos en el cerebro y es la responsable de evitar que no lleguen las bacterias. Así como nos protege, evita que la enzima llegue al sistema nervioso central.
-¿Hay consecuencias en caso de suspenderse la dosis?
-Se debe iniciar como si no lo hubiese recibido.
Lara posee el mayor número de casos con Hurler I
Cuando llegó el medicamento a Venezuela, el estado Lara fue el primero en recibirlo, porque existía el núcleo más grande de MPS I en el país, incluso, en el mundo. “En ese momento habían 20 historias de MPS I, cuando la frecuencia mundial era de 1000 personas recibiendo el tratamiento. Nosotros teníamos una monstruosidad comparado con todo lo que estaba ocurriendo a nivel internacional. Pero, eso ha bajado mucho”, informó el doctor Estrada. ¿Pero por qué Lara ocupaba el mayor número de casos? El especialista en genética de la UCLA, manifestó que se debe al efecto fundacional de Quíbor, donde hubo más pacientes.
“La MPS I es una enfermedad autosómica, eso tiene que ver con las Leyes de Mendel, cuando cruzamos los guisantes verdes con los guisantes amarillos puros, en la primera generación todos iban a salir amarillo, y salía amarillo porque este dominaba sobre el verde. De esa manera se comportan las enfermedades autosómicas recesivas como el Hurler. Cuando tengo un niño con Hurler I, los padres son portadores del mucopolisacaridosis, y ellos como pareja, cada vez que la señora esté en estado, va a tener un riesgo matemático de 25 %, o sea 1/4 de tener un niño con esas condiciones”, puntualizó.
-¿Por qué Quíbor?
-Aparentemente hubo en Quíbor el efecto fundador, una de las personas que llegó a fundar la población tenía el gen mutado a pesar de no tener la enfermedad, y como tuvo muchos hijos en esa población, se lo pasó a muchas personas. Quíbor muchas veces actuó como un aislado poblacional, porque cuando no existía la autopista centroccidental que va a Carora, no había buenas comunicaciones. Entonces la gente de Quíbor se unían entre ellos y al haber el gen mutado se podían unir dos personas que no sabían que eran familias. Eso ha cambiado toda vez la gente de Quíbor comenzó a relacionarse con personas de otros estados.[/vc_message]
Los medicamentos para las MPS en Venezuela llegaron en 2007 a Caracas. El primer paciente que recibió, por medios propios, el tratamiento fue el niño Humberto Da Silva con mucopolisacaridosis tipo II (Hunter), y luego de constantes luchas entre familiares de varios afectados, se logró que el Estado importara el medicamento a través de dos laboratorios.
Para la MPS II (Hunter) lo hizo mediante la empresa farmacéutica estadounidense, con sede fiscal en Irlanda, Shire (la misma que en enero de 2017 fue multada por 350 millones de dólares tras sobornos a médicos y recientemente fue vendida al laboratorio japonés Takeda); y en la MPS II, se utilizó como intermediario, a la trasnacional Genzyme Sanofi, con la cual se hicieron negocios desde 2008 para la importación de la enzima artificial Aldurazyme.
 Narvin Delgado, gerente de Comunicaciones de Genzyme Sanofi para Venezuela, manifestó que en la actualidad se canalizan algunas ayudas a través del International Charitable Access Program (ICAP) y con asesoría del doctor Manuel Andrade.
Narvin Delgado, gerente de Comunicaciones de Genzyme Sanofi para Venezuela, manifestó que en la actualidad se canalizan algunas ayudas a través del International Charitable Access Program (ICAP) y con asesoría del doctor Manuel Andrade.
Es de mencionar que el IVSS no ha entregado la data de los pacientes que requieren el medicamento. A continuación la entrevista que se le hizo a fin de conocer mayores detalles sobre el tema, sin embargo, en la misma fue imposible obtener por parte del laboratorio el monto que el Estado invierte, así como la data que ellos manejan. En dos interrogantes, Delgado respondió de una misma manera, evadiendo una información que de primera mano ellos dominan.
-¿Desde cuándo el Estado comenzó a importar el Aldurazyme a través de ustedes?
-Nuestro medicamento fue aprobado por la FDA (Administración de Alimentos y Medicamentos) y EMEA (Agencia Europea de Medicamentos) en el 2003. En Venezuela nuestra Laronidasa, inicialmente fue un medicamento de uso de servicio desde el 2011. Tiene su registro sanitario desde 2013 y está en el mercado venezolano desde diciembre del 2014.
-¿Cuánto fue la cantidad de medicamento para Hurler?
-Posología: Estos pacientes deben recibir su infusión de manera semanal. La dosis estándar es de 0,58 mg/kg. La presentación disponible viene en viales de 2,9 mg/5 cc.
-¿Cuánto fue la inversión, anual, o cómo, para conocer el costo del medicamento?
-En este caso creo que debes remitirte directamente a las autoridades pertinentes y solicitar esta información.
-¿Desde cuándo el Estado no envía la data para la compra del medicamento en MPS I?
-En este caso creo que debes remitirte directamente a las autoridades pertinentes y solicitar esta información.
-Entiendo que mediante el ICAP ustedes están ayudando a pacientes. Quisiera saber la data general de todos, en especial la de MPS I, si hay algunos que están en el programa
-Actualmente, brindamos soporte a los pacientes mediante el programa ICAP (International Charitable Access Program). Este es un programa global de Sanofi Genzyme que trabaja de la mano con instituciones gubernamentales, asociaciones de pacientes para brindarles tratamiento gratuito en aquellas patologías de depósito lisosomal, en las cuales tenemos opciones terapéuticas.
Este programa está regido por un marco referencial de Sanofi Genzyme global, en el que la población elegible para optar de este beneficio son los pacientes pediátricos menores a 5 años de edad que tengan el diagnóstico con patología de MPS tipo I, en la variante Hurler. Cada caso es sometido al análisis de una junta médica multidisciplinaria independiente e internacional que, finalmente, decide la incorporación del paciente al programa.
Te invitamos a acudir ante las autoridades sanitarias y solicitar la data general de pacientes.









